Preprocessing and averaging of single-channel NIRS data
Introduction
This tutorial demonstrates how to analyze a functional near-infrared spectroscopy (fNIRS) dataset focussing on one single channel. The goal is to introduce the basic fNIRS analysis features of FieldTrip on Artinis NIRS data. You can find details on the Artinis recording and analysis software here.
By the end of this tutorial, you will be able to read in fNIRS data, segment it and apply different pre-processing steps. This tutorial thus also serves as a kind of general introduction into the basic fundamentals of FieldTrip. You will also learn how to create basic visualizations of the data such as plotting single traces or topographic mapping. Finally, in this tutorial you will compute time-locked averages from the segmented data.
This tutorial does not show how to deal with bad channels as it only operates on single channel data. You can find more information about how to remove bad channels and generally how to analyise multiple channels in the Preprocessing and averaging of multi-channel NIRS data tutorial.
Background
NIRS is an abbreviation of Near-InfraRed Spectroscopy, which is a method to measure the level of oxygenation in the blood flowing through the tissue on which the sensors are placed. It relies on the fact that oxygenated hemoglobin has a different light absorption spectrum than de-oxygenated hemoglobin and hence it has a different color so you will. Most NIRS systems emit light of 2 wavelengths, typically around 700 and 850 nm, at every source optode and some systems use three wavelengths.
The detector optode is placed a few centimeters away from the source optode. Together, the source and detector optode make up a channel, but keep in mind that the system will always have 2 spatial locations for that one channel (most of us assume the recorded activity comes from a location mainly in the middle of the source and the detector) and 2 light wavelengths. The idea is that the light is scattered through the tissue and a small portion of the light will be detectable at the surface of the tissue by means of light detectors. The intensities of the light detected at the skin fluctuate systematically with changes in the oxygenated and de-oxygenated hemoglobin concentrations present in the tissue that lies between the source and detector optode. Due to the (more or less random) scatter, most light that arrives at the detector has only traveled through the surface of the tissue and only some small portion has traveled through an area a bit deeper below the surface. Typically, it is assumed that the light detected at the skin has traveled through a tissue volume that has a banana shape when depicted from a side-view. If the source and the detector are placed further apart, the light detected can potentially have traveled deeper through the tissue. Practically, there are limitations, because the portion of light that arrives at the detector falls off dramatically with increasing source-detector separations. When measuring changes in hemoglobin in the brain, the skull functions as an important barrier, as much light is absorbed by the bone. For that reason, scanning deeper in the brain is only feasible in young infants who still have a thin skull.
The concentration of oxy- and deoxygenated hemoglobin can not be assessed in absolute terms, which means that all measures will reflect relative changes. Hence, like EEG and MEG, a baseline period is required. This can either be time-locked to the events that are provided to the participant (trial-by-trial baselines) which implies functional NIRS (fNIRS) or measured in a somewhat longer period prior or after the recording of interest (NIRS).
The recorded and later processed fNIRS signal is comparable to fMRI in that sense that a hemodynamic response function (HRF) can be obtained, but with a higher temporal resolution. It is therefore, at least in theory, possible to measure the initial dip in the curve. In contrast to fMRI, the spatial resolution however is much poorer, and typically, we mainly record from the surface of the brain. As a result, plotting will often resemble EEG. The temporal resolution of EEG is of course much higher, but, the positive side of fNIRS is that the measured signal can only stem from the region in-between the source and detector, which need not be the case in EEG due to smearing. As such, fNIRS is a promising method for measuring brain activity in populations that cannot be tested in an fMRI scanner (think of young infants or people with cochlear implants who would otherwise display larger artifacts in regions that might be of critical interest), and/or for tasks that require more movements than the scanner would allow. Do keep in mind though that blood pressure can change with your task and thereby affects the measured signal as well.
The dataset used in this tutorial
In this dataset the motor cortex was probed using an Oxymon MK III system of Artinis Medical Systems. The optodes were placed over the motor cortex, and subsequently the subject was asked to perform finger tapping. Alongside the recordings from the brain, we recorded an event channel with annotations describing what the participant was doing. In the event channel, an ‘A’ was recorded at the moment the participant started finger tapping, and a ‘B’ was recorded at the moment the participant stopped tapping. This motor task was repeated for a total of 12 times and all data was saved, including the event channel, in an .oxy3 file.
The data used in this tutorial is available from our download server on https://download.fieldtriptoolbox.org/tutorial/nirs_singlechannel/. Specifically, you should download motor_cortex.oxy3 and optodetemplates.xml. For the XML file please right-click and use the save-as option.
Procedure
Analyses can be conducted in many different ways and in different orders, depending on the data and on the experimental design. We will first introduce you to a standard order of analysis steps, which you can subsequently try out step-by-step in this tutorial.
The following steps provide a good standard approach for analyzing fNIRS data, see Figure 1 for an overview:
- read continuous data
- (optionally) trim the beginning and the end of the recording if these are noisy and not of interest
- remove bad segments in the channels of interest, e.g., due to motion artifacts
- transform optical densities to changes in oxyhemoglobin (oxyHb) and deoxyhemoglobin (deoxyHb) concentration
- separate functional from systemic responses; which can be done using either one of, or a combination of
- filtering; i.e. temporal processing
- subtracting reference channel; i.e. spatial processing
- anti-correlating oxyHb/deoxyHb-traces per channel
- define epochs corresponding to the trials in the experimental task
- average the data over trials and visualize
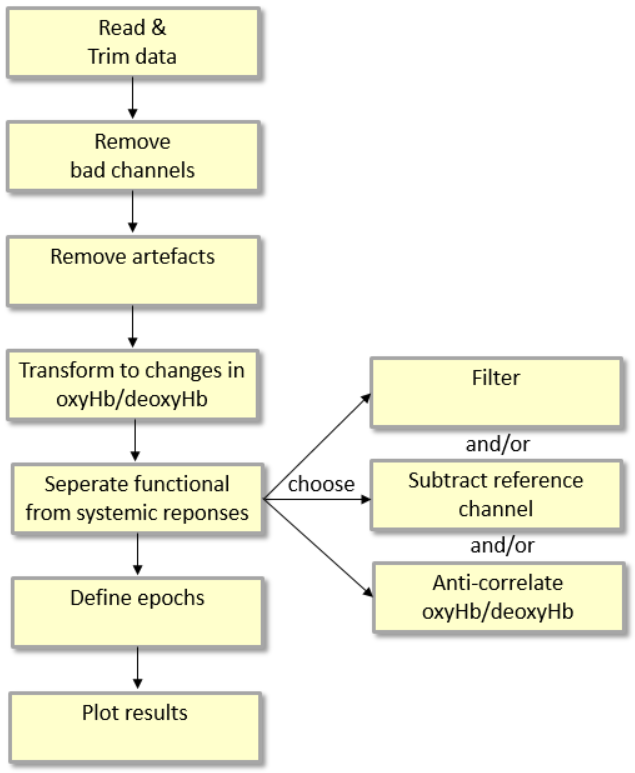
Figure 1: Overview of a standard fNIRS analysis procedure.
Getting Started
The dataset that we would like to analyze is called ‘motor_cortex.oxy3’. First, we need to point FieldTrip to this file. Note that FieldTrip does not feature a graphical-user interface, but instead requires you to script your way to the end goal. To let MATLAB know that we want to use FieldTrip and it’s file locations, add the FieldTrip folder to your path (without subfolders) and then execute the ft_defaults function by typing:
ft_defaults
In general, FieldTrip is keeping all input arguments to a function stored together in a local variable called ‘configuration’ or ‘cfg’. Although MATLAB variables could take any name, for convenience we will keep on using the name ‘cfg’ for this. FieldTrip functions are designed to each represent one step in your analysis pipeline. One (crucial) step in your analysis pipeline is to read in and preprocess the data. For this, we will make use of the FieldTrip function ft_preprocessing. You can check out the help of the preprocessing function by typing
help ft_preprocessing
It might also be helpful to check out the code to obtain an understanding of what the function is about. As FieldTrip is an open source toolbox, you can always have a look at the code details by typing:
edit ft_preprocessing
If this is your first time using FieldTrip you might also want to have a look at the introduction to the toolbox and MATLAB tutorial.
Read & trim data
For now, however, let’s take a moment and read the help only and neglect the code. So, it seems that ft_preprocessing would like to see our filename in the variable cfg.dataset. So, let us give FieldTrip this information. Best is to open up the editor and put all lines of code in there. It is also good practice to initialize the cfg-variable each time you want to start a new step in your processing pipeline.
cfg = [];
cfg.dataset = 'motor_cortex.oxy3';
Normally you would specify the full path, including the drive letter. This makes sure that your code will execute independent from your current MATLAB location. It is good practice to keep your original raw data, your processed data and your scripts in three separate locations. In this case we will work with the data in the present working directory.
Let us try to execute ft_preprocessing now as was specified in the help:
[data] = ft_preprocessing(cfg);
When working with .oxy3 files, the optode template containing the layout of the optodes needs to be loaded. If MATLAB cannot find the template file on your path, it will pop up a graphical user interface dialogue asking you to locate the XML file. By default this file is located in C:\Program Files (x86)\Artinis Medical Systems BV\Oxysoft 3.0.103. The file to select is optodetemplates.xml. However, given that FieldTrip often will search for this function, it is best to copy this function to the same folder as where your MATLAB analysis script and/or your fNIRS are data stored. For information on how to choose the optimal template for your experiments, please see this blog post on the Artinis webpage.
FieldTrip is finished when you see something like this on the screen
>> the call to "ft_preprocessing" took 9 seconds
There are more options to specify when reading in your data, such as cfg.padding, cfg.padtype and cfg.continuous. Those options have default values or are determined automatically, so we ignore them here but you can find all of them in the help documentation.
Through the option cfg.trl you can specify which trials should be read in. For now we will read in all trials, but we will make a selection later on.
Let us take a look at what just happened. If you observed your workspace closely, you will have noticed that a new variable was added called ‘data’. This is the same name that we put just in front of ft_preprocessing. Had you written [something], then the variable ‘something’ would have been added to your workspace.
If preprocessing was done as described, the data will have the following fields
data =
hdr: [1x1 struct]
label: {48x1 cell}
time: {[1x4462 double]}
trial: {[48x4462 double]}
fsample: 5
sampleinfo: [1 4462]
opto: [1x1 struct]
cfg: [1x1 struct]
Let us go through these one-by-one. The field ‘hdr’ contain all top-level information about your data, like for example the original sample rate, the number of channels etc. So all information that was potentially available at the time you read in the dataset. The field ‘label’ lists the name of all channels that you decided to read in. Note that you have told ft_preprocessing to just read-in, by default, all channels as you haven’t specified a subset. As you can see, there are 48 labels, and as the measurement consisted of 2 wavelengths per channel, this represents 24 channels. You also read in a set of ADC-channels, these will be ignored for now (these contain the triggers of the oxymon file, hence, this is NIRS acquisition hardware specific). The next field is called ‘time’ and represents the time axis of the dataset. The field ‘trial’ contains the data of all your channels. It is called ‘trial’ because usually, data is cut in segments corresponding to the different trials in the experiment. To start off with the analysis here, we read in all available data as a single segment/trial. The field ‘fsample’ describes the sampling rate of the data in the ‘trial’-field. The field ‘sampleinfo’ describes the sample numbers of each trial with respect to the original measurement on disk. The field ‘opto’ contains information about the composition of the channels and optodes, such as what wavelengths were used, the position of the optodes, how the optodes were combined to form the channels, etc. Finally, the field ‘cfg’ is the same cfg that we have just used, extended by some default values. This way, we can always trace back what has actually happened to our data. But more about that later.
Let us dive deeper into our data for now. For having a quick look at our data, we can use the function ft_databrowser . The databrowser is much more than a simple ‘data browser’, but we will utilize this functionality for our purpose at the moment. Of course, we could have a look at how to call the databrowser (help ft_databrowser), but a good guess is always to use FieldTrip functions as ft_functionname(cfg, data). We can keep the cfg empty to start with, and then see if this works. We add ‘ylim = ‘maxmin’’ to the configuration to adjust the y-axis such that the lowest values in the graph are determining the lowest point on the y-axis, and the largest values in the graph determine the highest point on the y-axis.
cfg = [];
cfg.ylim = 'maxabs';
ft_databrowser(cfg, data);

Figure 2: Display of raw data in the databrowser.
Using ft_databrowser, you can also cut out pieces of your data that you do not need. For instance, if you have started the recording while putting the optodes in place, you will probably have a chunk of data at the start of the recording that you don’t need and which contains very high (not brain-related) values that rapidly fluctuate. It is useful to cut these pieces out (trimming). In the tutorial dataset, this is not needed, but see here for an illustration of how trimming works within ft_databrowser.
Additionally, we’ll from here select just one pair of channels, to reduce the complexity for those new to fNIRS analyses.
cfg = [];
cfg.ylim = 'maxmin';
cfg.channel = {'Rx4b-Tx5 [860nm]', 'Rx4b-Tx5 [764nm]'}; % you can also use wildcards like 'Rx4b-Tx5*'
ft_databrowser(cfg, data);
Figure 3: Display of one pair of channels (two wavelengths) in the databrowser.
Exercise 1
Take a moment to familiarize yourself with the user-interface. Change the horizontal and vertical scales until you can see the heartbeat signal in the selected channels. Tip: A time slice of something between 10 to 20 seconds is optimal. Picking up the heartbeat in the NIRS measurement is a sign of good data quality, if the heartbeat cannot be detected in the data, contact with the skin must have been poor.
Remove artifacts
There are several ways to remove aspects of the data that are not of interest. That is, fNIRS data not only represents changes in oxyhemoglobin and deoxyhemoglobin concentrations, but contains, amongst others, also other physiological signals, random noise and variations stemming from the measurement environment. One prominent issue is motion artifacts, which are produced by temporary changes in the contact between optode and skin, often caused by movements of the head. Typically, one finds these motion artifacts in all channels simultaneously, but it could also be that just one channel or a few channels were affected, for instance if the participant moves the mouth. Short, unexpected peaks in the data are considered to stem from motion. You can detect and remove these artifacts for instance through ft_artifact_zvalue.
You can specify some preprocessing options to become sensitive to the artifacts and a z-value cut-off like this:
cfg = [];
cfg.artfctdef.zvalue.channel = {'Rx4b-Tx5 [860nm]', 'Rx4b-Tx5 [764nm]'};
cfg.artfctdef.zvalue.cutoff = 5;
cfg.artfctdef.zvalue.hpfilter = 'yes';
cfg.artfctdef.zvalue.hpfreq = 0.1;
cfg.artfctdef.zvalue.rectify = 'yes';
cfg.artfctdef.zvalue.artpadding = 2;
% cfg.artfctdef.zvalue.interactive = 'yes'; % the interactive display makes more sense after segmentating data in trials
[cfg, artifact] = ft_artifact_zvalue(cfg, data);
You will see that FieldTrip identified 8 artifacts through this procedure. Note that these are only identified, they are not yet removed; we will keep them in memory and call ft_rejectartifact after filtering segmenting the data into epochs.
Exercise 2
Play around with the cut-off z-value. You can do this by running the artifact rejection in interactive mode by adding
cfg.artfctdef.zvalue.interactive = 'yes';
before you run ft_artifact_zvalue. In the interactive mode, you can interactively change the threshold to see which parts of the data would be rejected, the rejected bits are marked in red. You can call the code multiple times, changing the filter settings, to try to become more sensitive for the movement artifacts.
What is the optimal threshold to get rid off short-lived transient peaks?
Transform to changes in oxyHB/deoxyHB
You might have noticed that you were looking at optical density (OD) values rather than at oxygenated and deoxygenated hemoglobin concentrations, because the channel labels mention the wavelengths. The optical density values directly relates to the light intensity that picked up by the the optodes. We can transform our data to concentrations using ft_nirs_transform_ODs. One of the choices to make when using ft_nirs_transform_ODs is the differential path-length factor or DPF, which differs depending on the age of the participant and the tissue type under investigation (e.g., when analyzing changes in blood oxygenation in muscle rather than brain).
cfg = [];
cfg.dpf = 5.9;
cfg.channel = {'Rx4b-Tx5 [860nm]', 'Rx4b-Tx5 [764nm]'};
data_conc = ft_nirs_transform_ODs(cfg, data);
Exercise 3
Check out the data again! As expected, the selected channel, in which you were able to see a clear heartbeat in the raw signal, also shows a clean signal once transformed to oxy- and deoxyHb values, right?
Separate functional from systemic responses
Apart from motion artifacts, which appear as spikes in the data, fNIRS measurements also contain systemic responses, which we are not interested in and want to filter out of our data. There are multiple ways of extracting the functional response (the haemodynamic response of the brain), but for now, we take a simple but relatively effective approach, namely by applying a bandpass filter to eliminate signal contributions outside a user-defined frequency band that captures the dynamics in the hemodynamic response of the brain. Due to the experimental task and the haemodynamic response function, we expect activity below 0.1 Hz (corresponding to fluctuations of ~10 seconds). Additionally we set the lower range to 0.01 Hz (fluctuations of ~100 seconds) to eliminate slow drifts.
cfg = [];
cfg.bpfilter = 'yes';
cfg.bpfreq = [0.01 0.1];
data_filtered = ft_preprocessing(cfg, data_conc);
Define epochs of interest
Because we are interested to find whether the brain responds specifically to the events that took place during the experiment, we want to focus our analyses on the timewindows in which the events took place (these timewindows are often called epochs or trials). More specifically, we want to investigate whether there is increase in oxygenation in the channel of interest timelocked to the experimental event. In the current dataset, the events are fingertaps of the participant. To be able to analyze these specific event-related responses, we have not only recorded the NIRS signal, but also when which event happened, stored in a so-called trigger channel. The triggers indicate when what happened in the experiment.
Earlier, we have read in all epochs by not specifying cfg.trl during our previous call to ft_preprocessing. In the help documentation cfg.trl points to ft_definetrial, which we will now use to define our trials. So let us have a look at the function
help ft_definetrial
Let us assume for now that we have no clue about the triggers in the data. We will thus now utilize a more-or-less hidden functionality to retrieve this information.
cfg = [];
cfg.dataset = 'motor_cortex.oxy3';
cfg.trialdef.eventtype = '?';
The question mark indicates that we are not sure about the event triggers, and the function ft_trialfun_general will thus output all events that are found in the dataset. Now, we can call ft_definetrial
ft_definetrial(cfg);
In the command window, you will see that 24 events were found, and two types of events were used, namely event ‘A’ and event ‘B’. In case you recorded your files with the Artinis Oxymon system, and you did not add events during the measurement, you can include them here, see FAQ.
Furthermore, note that we do not define the output variable ‘cfg’ now, as we are not interested in the output. The variable ‘cfg’ will thus stay unchanged. We want to have a look at epochs/trials starting 10 second pre-stimulus (before event ‘A’) and ending 35 seconds after ‘A’ (post-stimulus). So we can now define our trials and subsequently use this to call ft_preprocessing:
cfg.trialdef.eventtype = 'event';
cfg.trialdef.eventvalue = 'A';
cfg.trialdef.prestim = 10;
cfg.trialdef.poststim = 35;
cfg = ft_definetrial(cfg);
cfg.channel = {'Rx4b-Tx5 [860nm]', 'Rx4b-Tx5 [764nm]'};
data_epoch = ft_redefinetrial(cfg, data_filtered);
We have now selected one pair of channels and cut the data in 12 trials. Check them out using the databrowser, but let us use some settings to make the plots look neater and also visualize the artifacts that we identified earlier:
cfg = [];
cfg.ylim = [-1 1];
cfg.viewmode = 'vertical';
cfg.artfctdef.zvalue.artifact = artifact;
ft_databrowser(cfg, data_epoch);
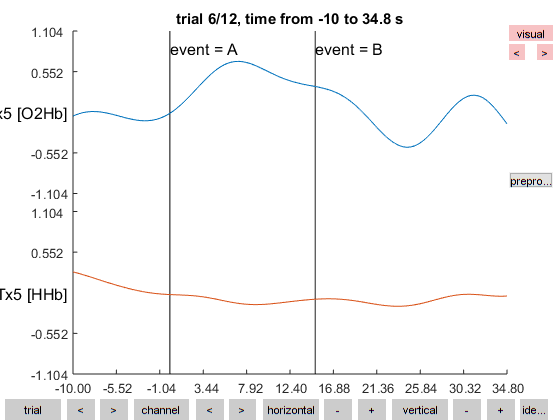
Figure 4: Databrowser showing the filtered data for one of the 12 trials.
The artifacts that we identified earlier are not so clearly visible any more due to the filtering. You can use similar code to look at the artifacts in the unfiltered data.
We can now remove the trials containing the artifacts that we determined earlier.
cfg = [];
cfg.artfctdef.zvalue.artifact = artifact;
cfg.artfctdef.reject = 'complete';
data_epoch = ft_rejectartifact(cfg, data_epoch);
So we pulled our data out of the measurement. The data looks crisps and clear.
Exercise 4
All signal values seem to be around the same values. Why could that be?
Exercise 5
You might want to perform an additional preprocessing step now. What steps do you consider useful? Check out the options in ft_preprocessing!
Timelockanalysis
We proceed by computing the average over trials using ft_timelockanalysis. Subsequently, we can use FieldTrip plotting function to look at the data, or simply use the MATLAB plot function for visualization.
cfg = [];
data_timelock = ft_timelockanalysis(cfg, data_epoch);
The output is the data structure data_timelock with the following field
data_timelock =
avg: [2x225 double]
var: [2x225 double]
time: [1x225 double]
dof: [2x225 double]
label: {2x1 cell}
dimord: 'chan_time'
opto: [1x1 struct]
cfg: [1x1 struct]
The most important field is data_timelock.avg, containing the average over all trials for each channel.
Below we plot the averaged O2Hb and HHb traces from A-10 seconds to A+35 seconds. IN line with fNIRS convention, O2Hb is coloured red and HHb is coloured blue.
time = data_timelock.time;
O2Hb = data_timelock.avg(1,:);
HHb = data_timelock.avg(2,:);
figure;
plot(time,O2Hb,'r'); hold on;
plot(time,HHb,'b');
legend('O2Hb','HHb'); ylabel('\DeltaHb (\muM)'); xlabel('time (s)');
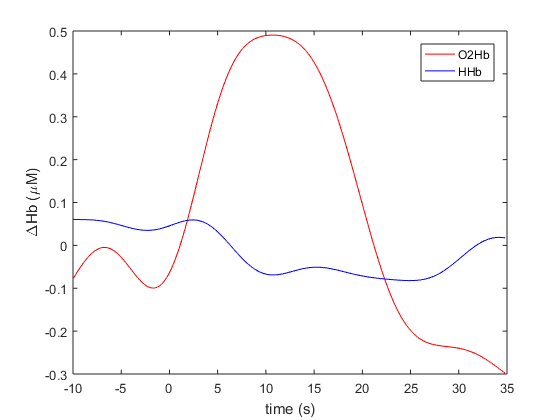
Figure 5: Averaged O2Hb and HHb traces. This figure closely resembles the text-book fNIRS model of cortical activation, which results from an increase in oxygen demand from the tissue instigating an increase in O2Hb due to neuro-vascular coupling as depicted by Scholkmann et al. in figure 5 of their 2014 review article.
Summary and conclusion
We explained the preprocessing steps for a single channel in an fNIRS dataset using FieldTrip. If you would like to read further on how to preprocess an fNIRS dataset with multiple channels, you can continue with the fNIRS multi-channel tutorial. When you have more questions about the topic of any tutorial, do not forget to check the frequently asked questions and the example scripts.
See also the other documentation that relates to fNIRS:
- Getting started with Artinis NIRS data
- Converting an example NIRS dataset for sharing in BIDS
- Getting started with Hitachi NIRS data
- Getting started with Homer
- Specifying the channel layout for plotting
- Analyzing NIRS data recorded during unilateral finger- and foot-tapping
- Using GLM to analyze NIRS timeseries data
- Creating a layout for plotting NIRS optodes and channels
- Preprocessing and averaging of multi-channel NIRS data
- Preprocessing and averaging of single-channel NIRS data
- Analyzing NIRS data recorded during listening to and repeating speech
- Getting started with NIRx NIRS data
- Getting started with Shimadzu NIRS data
- Getting started with SNIRF data
- Making a synchronous movie of EEG or NIRS combined with video recordings